

Introducing Evo, a long-context biological foundation model based on the StripedHyena architecture that generalizes across the fundamental languages of biology: DNA, RNA, and proteins. Evo is capable of both prediction tasks and generative design, from molecular to whole genome scale (over 650k tokens in length). Evo is trained at a nucleotide (byte) resolution, on a large corpus of prokaryotic genomic sequences covering 2.7 million whole genomes.
Evo is an OSS model built on the StripedHyena architecture, a deep signal processing architecture designed to improve in efficiency and quality over the prevailing Transformer. Evo-1 was collaboratively developed together by Together AI and the Arc Institute.
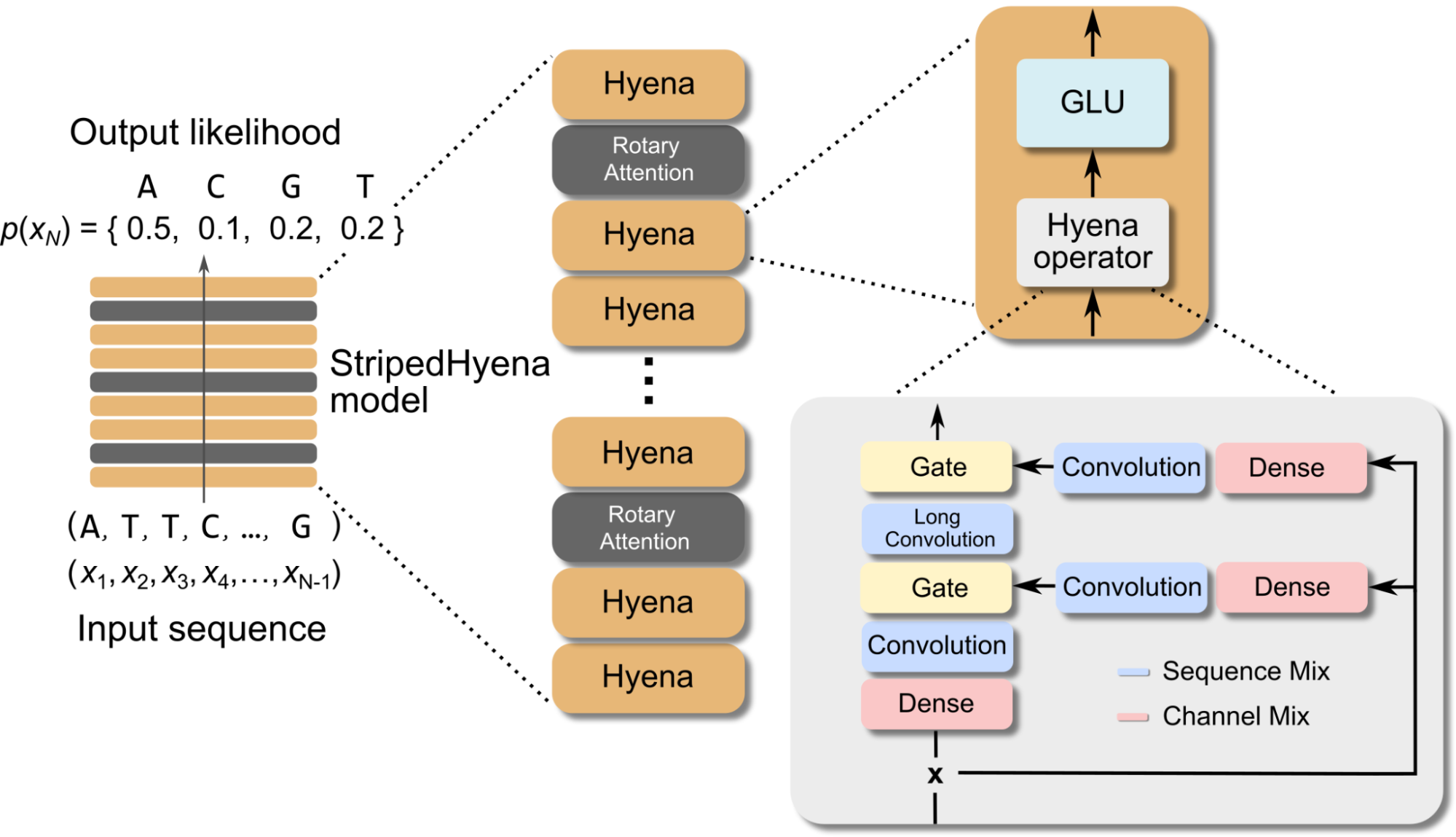
The model is available on HuggingFace, in this repository, via the Together API and Playground.! In addition to model weights, we are also excited to share intermediate checkpoints. We will release the training dataset (OpenGenome), consisting of 2.7M publicly available genomes from prokaryotes, in the coming days.
Read more in our paper.
Is DNA all you need?
In biology, everything starts with DNA. Genomes carry an entire set of DNA (the genetic code) to make a complete organism. Within them lies the result of generations of evolution, reflecting adaptations to constantly shifting environmental changes. Other complex biological languages emerge from this code, including proteins, the tiny molecular machines that make cells function, and RNA, which helps DNA transmit information and often helps proteins accomplish their functions. As multilayered as these languages seem, they are all unified in (our) genomes.
The emergence of AI foundation models has charted a promising path in biological sequence modeling, yet modeling at the whole-genome level has been out of reach for many methods. DNA sequences are extremely long (up to billions of nucleotides), and the sensitivity required to fully understand the effects of evolution (which occurs one nucleotide at a time), makes it a particularly challenging domain for large-scale pretraining. It’s unclear if AI models are able to learn such complex patterns. As a result, existing breakthroughs in modeling biological sequences with AI have instead focused on task-specific or single-modality capabilities
These challenges (and the fundamental question of whether DNA is all you need) motivated us to work on Evo. In particular, we wanted a foundation model that could integrate information over long genomic sequences while retaining sensitivity to single-nucleotide changes. A model that effectively learns over genomes could understand not only the individual DNA, RNA, and protein components, but also how these interact to create complex systems. This could accelerate our mechanistic understanding of biology and the ability to engineer life itself.
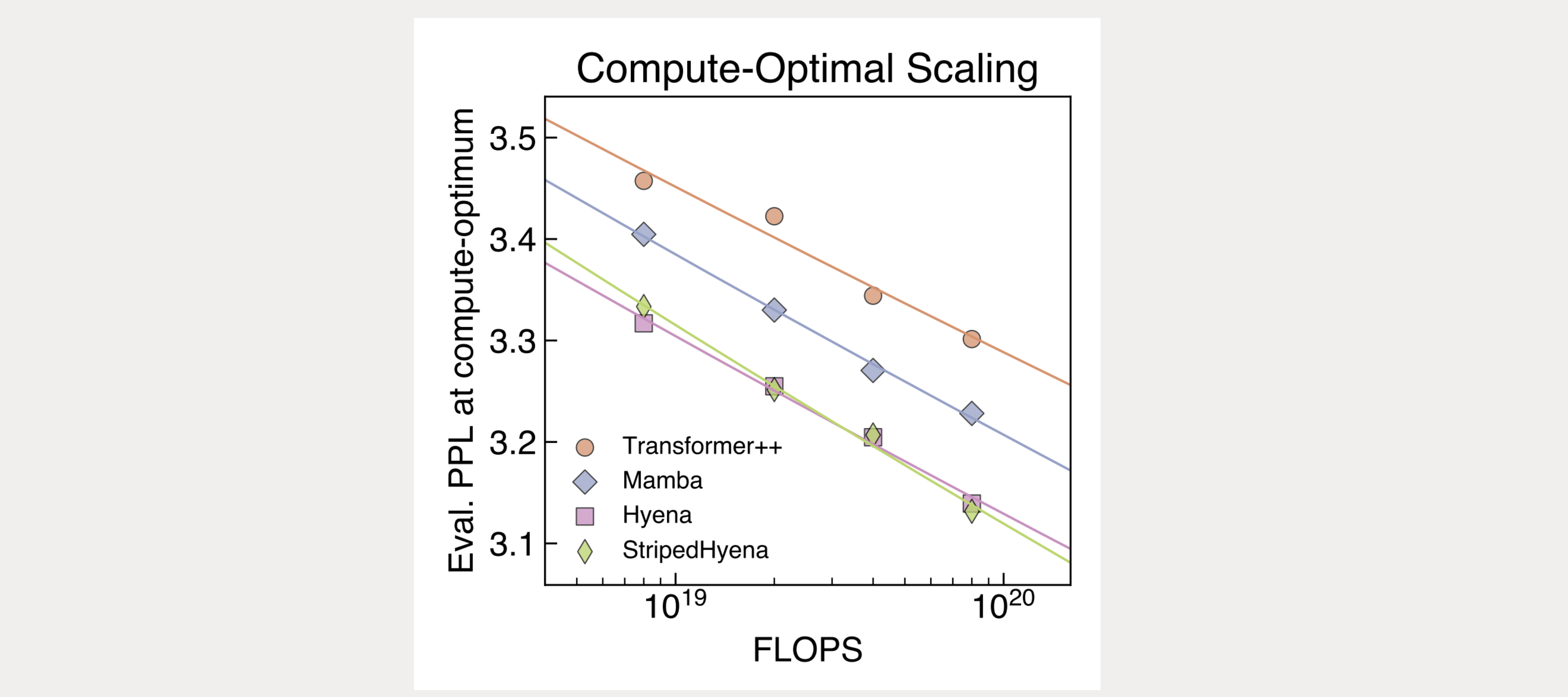
Demonstrating the first scaling laws on DNA pretraining
We carry out a first-of-its-kind scaling laws analysis on DNA pretraining, and find Transformer models do not scale as well when trained at single-nucleotide, byte-level resolution.
To overcome the challenges associated with sequence modeling at long sequence lengths and at byte-level resolution, we used the StripedHyena architecture. Evo achieves both long context and nucleotide resolution via our latest advances in architecture design, hybridizing rotary attention and hyena operators to efficiently process and recall patterns in long sequences.
Evo-1 capabilities
Zero-shot gene essentiality testing
Strikingly, Evo understands biological function at the whole genome level. Using an in silico gene essentiality test, Evo can predict which genes are essential to an organism’s survival based on small DNA mutations. It can do so zero-shot and with no supervision. For comparison, a gene essentiality experiment in the laboratory could require 6 months to a year of experimental effort. In contrast, we replace this with a few forward passes through a neural network.
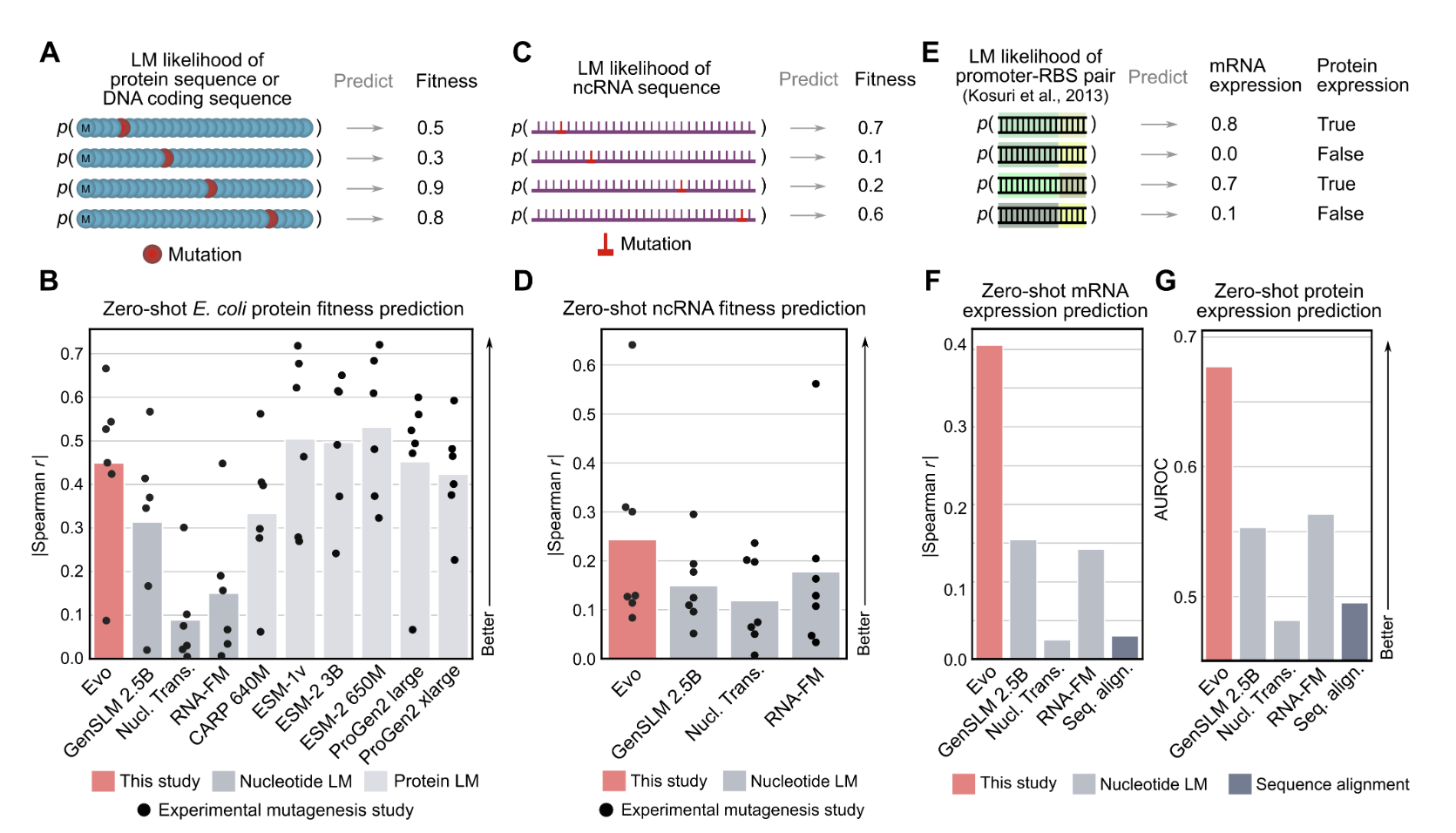
Zero-shot prediction across DNA, RNA, and protein modalities
Because Evo is trained on long genomic sequences that contain protein coding sequences, we tested whether the model would also learn the protein language well enough to perform zero-shot protein function prediction. Evo outperforms all other nucleotide models tested, including models explicitly trained only on protein coding sequences, and is even competitive with state-of-the-art protein language models, like ESM or ProGen. But there are more than just proteins in Evo’s genomic training data—there are ncRNAs and regulatory DNA sequences in genomes as well. Notably, we show that Evo enables zero-shot function prediction for ncRNA and regulatory DNA, as well, thereby spanning all three modalities of the central dogma.
CRISPR system generation
Right now, generative models for biology are mostly focused on a single modality—for example, only on proteins or on RNA. One of the key breakthroughs we highlight is that Evo can perform multimodal design to generate novel CRISPR systems, a task that requires creating large functional complexes of proteins and ncRNA, and is out of reach for existing generative models. Right now, generating new CRISPR systems requires searching through natural genomes for similar sequences that were literally taken from an organism. Instead, Evo enables a new approach to generating biological diversity by sampling sequences directly from a generative model, an exciting frontier for creating new forms of genome editing tools.
Genome scale generation
Evo can not only generate at the scale of multiple molecules (proteins and ncRNA), it has the potential to generate sequences at the scale of whole genomes. We can generate sequences of up to 650k on a single GPU. Generating sequences of this length benefits from both long context capabilities of the architecture, as well as from its efficient inference mode. When we sample sequences at this length with Evo, we find genomes that contain thousands of potential protein-coding sequences.
Generating sequences of this length benefits from both long context capabilities of the architecture, as well as from its efficient inference mode. We can generate sequences of up to 500k on a single GPU.
Safe and responsible development of Evo
Evo is the first of its kind to predict and generate DNA sequences at the whole-genome scale with single-nucleotide resolution. Future capabilities that emerge from large-scale DNA models like Evo also require additional work to ensure that these capabilities are deployed safely and for the benefit of humanity. In our paper, we provide an extended discussion on potential risks and precautionary measures.
Future plans
Evo marks a turning point in what we think is possible in modeling biological sequences, and beyond. We believe this technology has the potential to accelerate discovery and understanding in the sciences (such as biology, chemistry, or material science), as well as be applied to real-world problems including drug discovery, agriculture, and sustainability. Although the results show promising computational capabilities, further experimental validation is required for the generated sequences.
Foundation models are going to be increasingly important scientific tools. We look forward to training larger models, improving their generation capabilities, and expanding Evo pretraining to human genomes. We also want to increase the level of biological complexity learned by these models to make progress on fighting complex diseases and improving human health.
We believe foundation models are going to be increasingly important scientific tools. We look forward to contributing to the AI ecosystem in biology, by training larger models and expanding our support with dedicated playground features.
Acknowledgments
The full research team behind Evo: Eric Nguyen, Michael Poli, Matthew Durant, Armin Thomas, Brian Kang, Jeremy Sullivan, Madelena Ng, Ashley Lewis, Aman Patel, Aarou Lou, Stefano Ermon, Stephen Baccus, Tina Hernandez-Boussard, Chris Ré, Brian Hie, Patrick Hsu.